VASP如何计算单原子能量
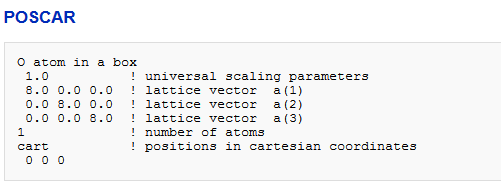
1、我们捂执涡扔使用的是一个带有单个原子的POSCAR文件。选择了足够大的晶格参数,从而使相邻细胞之间的原子之间不存在(相对较大)的相互作用。POSCARO atom in a box1.0 ! universal scaling parameters8.0 0.0 0.0 ! lattice vector a(1)0.0 8.0 0.0 ! lattice vector a(2)0.0 0.0 8.0 ! lattice vector a(3)1 ! number of atomscart ! positions in cartesian coordinates 0 0 0
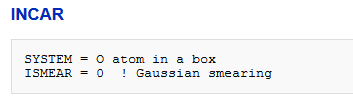
2、INCAR文件SYSTEM = O atom in a boxISMEAR = 0 !高斯近似
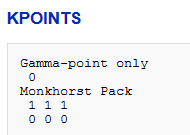
3、K点选取KPOINTSGamma-point only0怡觎现喾Monkhorst Pack1 1 10 0 0对于原子或分子来说,单k点就足够了。当更多的k点被使吹涡皋陕用时,原子间的相互作用(应该为零)可以更准确地描述。
4、计算结果如图所示,本文中使用的是VASP5.4.1版本,初始电荷与孤立重叠的原子(POTCAR文件)的电荷有关。在前4个步骤中,电荷保持不变,之后开始变化
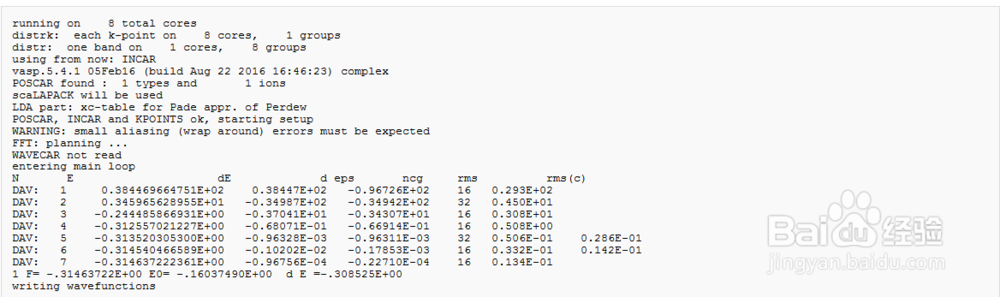
5、OSZICAR and stdout 文件中符号的含义N :迭代次数E : 总能量dE :总能量的变化d : 本征值的变化,每个K点对应很多个能级,每个能级对应的能量就是本征值
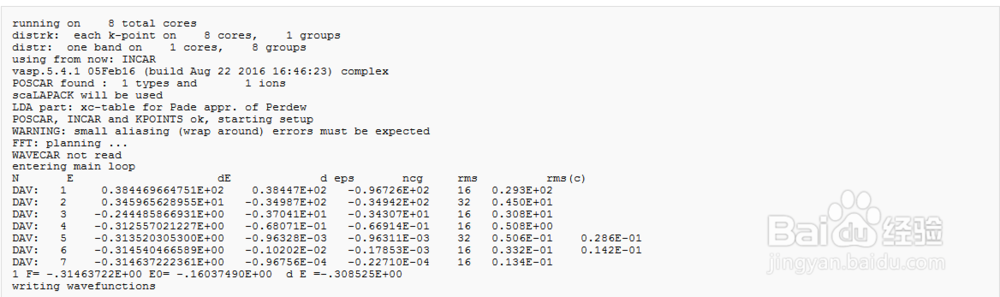
6、OUTCAR文件包含如下及部分:读取INCAR, POTCAR, POSCAR相邻原子的距离,分析对称性工作信息晶格,K点,位置的信息基组的信息,平面波的数量全局赝势能每一个离子步的信息耗时和能量信息

声明:本网站引用、摘录或转载内容仅供网站访问者交流或参考,不代表本站立场,如存在版权或非法内容,请联系站长删除,联系邮箱:site.kefu@qq.com。
阅读量:24
阅读量:88
阅读量:39
阅读量:28
阅读量:67